Effortless documentation starts here
Centralize your team’s knowledge, sync with your codebase, and create beautiful documentation your customers and teams will love
Effortless documentation starts here
Centralize your team’s knowledge, sync with your codebase, and create beautiful documentation your customers and teams will love
Effortless documentation starts here
Centralize your team’s knowledge, sync with your codebase, and create beautiful documentation your customers and teams will love
Effortless documentation starts here
Centralize your team’s knowledge, sync with your codebase, and create beautiful documentation your customers and teams will love

Trusted by technical teams at companies of all sizes





Trusted by technical teams at companies of all sizes
Trusted by technical teams at companies of all sizes
Trusted by technical teams at companies of all sizes
Upgrade to smarter
technical documentation

Create access-protected docs
With visitor authentication you choose who can access your docs — so you can keep sensitive information away from competitors and hackers. Simply integrate with your auth platform and only authorized users can view your documentation.

Create access-protected docs
With visitor authentication you choose who can access your docs — so you can keep sensitive information away from competitors and hackers. Simply integrate with your auth platform and only authorized users can view your documentation.
Create access-protected docs
With visitor authentication you choose who can access your docs — so you can keep sensitive information away from competitors and hackers. Simply integrate with your auth platform and only authorized users can view your documentation.

A workflow you know
GitBook’s branch-based workflow will be instantly familiar if you’ve used GitHub or GitLab. Open a change request to edit a published or locked page, request a review, then merge.


A workflow you know
GitBook’s branch-based workflow will be instantly familiar if you’ve used GitHub or GitLab. Open a change request to edit a published or locked page, request a review, then merge.
A workflow you know
GitBook’s branch-based workflow will be instantly familiar if you’ve used GitHub or GitLab. Open a change request to edit a published or locked page, request a review, then merge.
A workflow you know
GitBook’s branch-based workflow will be instantly familiar if you’ve used GitHub or GitLab. Open a change request to edit a published or locked page, request a review, then merge.

Write better with AI
GitBook AI can fix grammar and spelling errors, explain technical jargon, edit your tone, translate your docs, or even write a first draft for you. All without leaving your GitBook tab.
Write better with AI
GitBook AI can fix grammar and spelling errors, explain technical jargon, edit your tone, translate your docs, or even write a first draft for you. All without leaving your GitBook tab.
Write better with AI
GitBook AI can fix grammar and spelling errors, explain technical jargon, edit your tone, translate your docs, or even write a first draft for you. All without leaving your GitBook tab.

Editing tools your technical writers will love
Our block-based editor lets you add and rearrange your content as you go — including code blocks and tables. Plus it has Markdown support for fast editing.

Editing tools your technical writers will love
Our block-based editor lets you add and rearrange your content as you go — including code blocks and tables. Plus it has Markdown support for fast editing.
Editing tools your technical writers will love
Our block-based editor lets you add and rearrange your content as you go — including code blocks and tables. Plus it has Markdown support for fast editing.
Editing tools your technical writers will love
Our block-based editor lets you add and rearrange your content as you go — including code blocks and tables. Plus it has Markdown support for fast editing.
Keep your docs and codebase in sync
With our GitHub and GitLab integrations, you can set up a frictionless two-way sync with a Git branch. Make changes to one, and the other updates automatically. So whether it’s your engineers or your technical writers making an edit, you know everything will stay up to date.
Sign up with GitHub
Keep your docs and codebase in sync
With our GitHub and GitLab integrations, you can set up a frictionless two-way sync with a Git branch. Make changes to one, and the other updates automatically. So whether it’s your engineers or your technical writers making an edit, you know everything will stay up to date.
Sign up with GitHub
Keep your docs and codebase in sync
With our GitHub and GitLab integrations, you can set up a frictionless two-way sync with a Git branch. Make changes to one, and the other updates automatically. So whether it’s your engineers or your technical writers making an edit, you know everything will stay up to date.
Sign up with GitHub

Create and maintain
rich, interactive docs

Effortless API documentation
Create detailed, interactive API docs that your users and customers will love — or generate them automatically from API definition files.

Effortless API documentation
Create detailed, interactive API docs that your users and customers will love — or generate them automatically from API definition files.
Effortless API documentation
Create detailed, interactive API docs that your users and customers will love — or generate them automatically from API definition files.

Collaboration leads to better documentation
In GitBook, you can collaborate using a branch-based workflow. Request a review of your changes, thread comments, and keep a full version history for every page. Or make edits alongside others in real-time.

Embed code sandboxes, demos, and more
GitBook’s block integrations let you embed all kinds of content into your pages — from live sandbox environments to interactive demos and Figma files.

Say goodbye to outdated documentation
GitBook’s AI content audits help you find and fix conflicting information fast. While user scores and search analytics help you see what your users like — and what needs more work.
Alpha

Search made simple
GitBook AI constantly indexes your content. So all you have to do is ask it a question — it’ll scan your documentation and summarize an answer in seconds.



Integrate with your stack
Install one of our verified integrations, or build your own with our API. Because a great knowledge management system should work with everything you use on a daily basis.

Ready to scale up?


Permission to grow
Stay in control as your team grows. With tiered permission controls, you chose who can see and edit your documents — making member management simple.

SSO and SAML
Make your team’s logins simpler and more secure with SSO. Sign in using an authorized email domain, or through a SAML 2.0 identity provider of your choice.

Security. At our foundations.
Your data security is our top priority. We have SOC 2 and ISO 27001 certification, and we’re GDPR compliant. So your can rest assured that your data is safe.
Ready to scale up?
Permission to grow
Stay in control as your team grows. With tiered permission controls, you chose who can see and edit your documents — making member management simple.
SSO and SAML
Make your team’s logins simpler and more secure with SSO. Sign in using an authorized email domain, or through a SAML 2.0 identity provider of your choice.
Security. At our foundations.
Your data security is our top priority. We have SOC 2 and ISO 27001 certification, and we’re GDPR compliant. So your can rest assured that your data is safe.
Ready to scale up?
Permission to grow
Stay in control as your team grows. With tiered permission controls, you chose who can see and edit your documents — making member management simple.
SSO and SAML
Make your team’s logins simpler and more secure with SSO. Sign in using an authorized email domain, or through a SAML 2.0 identity provider of your choice.
Security. At our foundations.
Your data security is our top priority. We have SOC 2 and ISO 27001 certification, and we’re GDPR compliant. So your can rest assured that your data is safe.

Create, search and manage your knowledge at scale. Effortlessly.
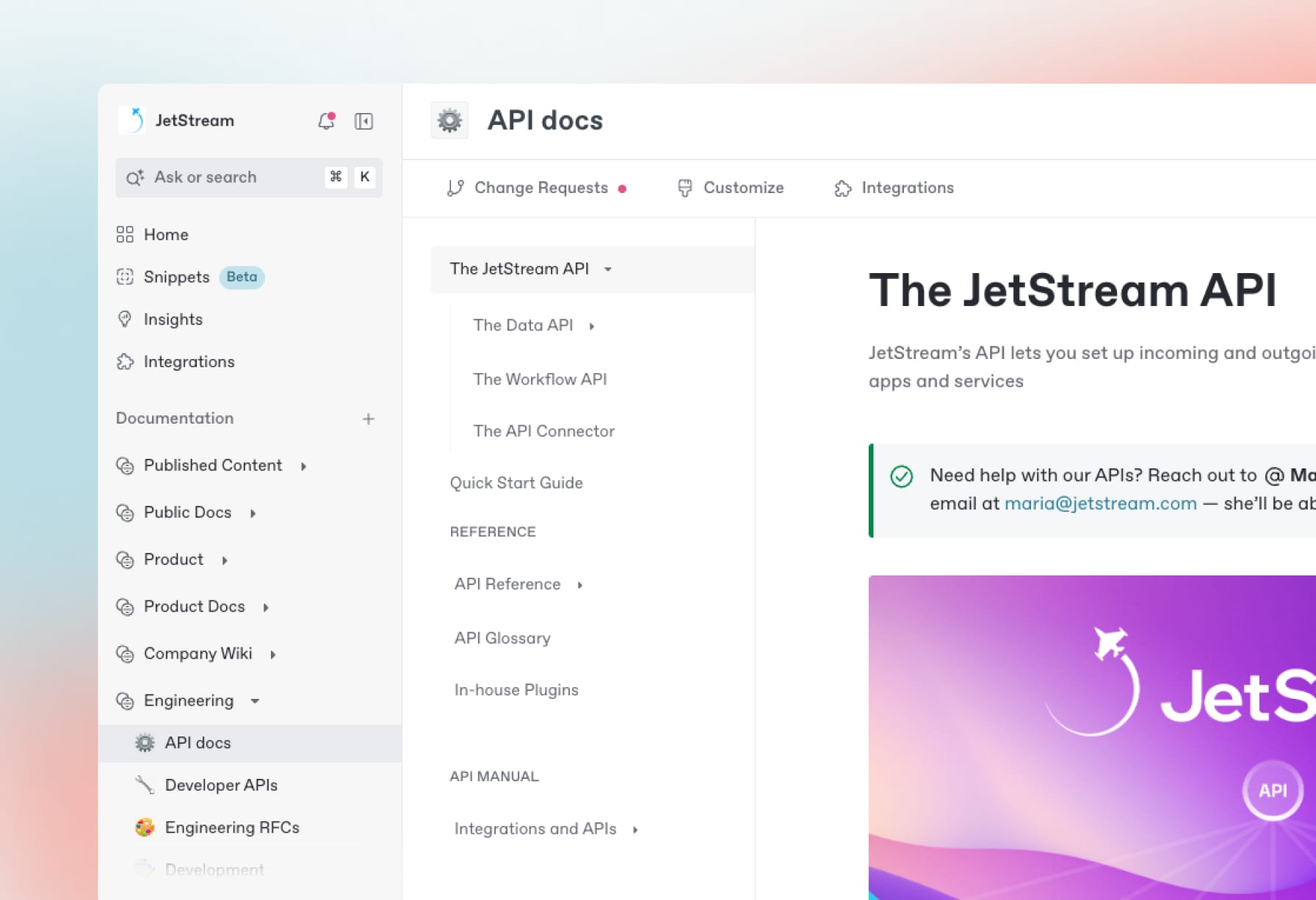
Create, search and manage your knowledge at scale. Effortlessly.
Create, search and manage your knowledge at scale. Effortlessly.
Create, search and manage your knowledge at scale. Effortlessly.